
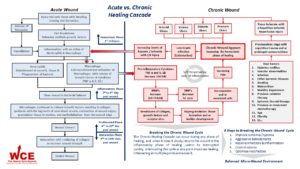
Acute wound healing is an orderly and efficient process which is characterized by four distinct, but overlapping phases: hemostasis, inflammation, proliferation and remodeling. In non-healing chronic wounds this efficient and orderly process has been lost due to the absence of the hemostasis phase, causing these ulcers to become locked into a state of chronic inflammation that is self-sustaining and characterized by abundant neutrophil infiltration with associated reactive oxygen species and destructive enzymes. Healing will only occur after this prolonged inflammatory phase is broken and the wound’s micro-wound environment is once again in balance thus allowing the wound to proceed through the remaining stages.1,88 At any point along the healing continuum a wound can shift back into the inflammatory phase of healing. When this happens, if the inflammatory cycle is broken quickly, the wound will continue to move forward along the healing continuum. If not, the wound will slowly return to a prolonged self-sustaining inflammatory phase and all progress will be lost.1,13,14,88
To develop an effective treatment regimen, a clearer understanding of the underlying pathology that results in a persistent cycle of inflammation and repetitive tissue damage is required. By bringing together relevant science and quality research, we aim to provide basic insight into the effects of this prolonged inflammatory phase of chronic wound healing.
The single most important aspect about chronic wounds is the absence of the hemostasis phase of healing. The sequence of events that is normally initiated by hemostasis, clot breakdown with the subsequent release of growth factors is absent and has a tremendous impact on healing which is the starting point of a prolonged inflammatory phase, leading to chronic non-healing wounds.1,13,14,88
Many researchers have suggested that repetitive ischemic-reperfusion injury and local tissue ischemia induce chronic inflammation in peri-ulcerative tissue and are the leading precursors of chronic non-healing wounds.2-14 Ischemic-reperfusion injury is a common medical condition that is considered to be the underlying medical condition in many cardiovascular and inflammatory diseases.15 The pathological features of an ischemic-reperfusion injury include accumulation of leukocytes in blood vessel lumens and leukocyte extravasation, elevated expression of inflammatory cytokines with tissue destruction proteinases, and fibrosis of microvessels, which are all features seen in other types of ischemia-reperfusion injury.7, 15 The four primary categories of chronic wounds are: pressure, diabetic, arterial, and venous ulcers, which are the overwhelming majority of chronic wounds and all share these common causative factors.16
Local tissue ischemia and repetitive reperfusion injury impairs both the microcirculation and macrocirculation, and affects small vessel hypertension which is the initial starting point of inflammation within tissue. Compounding this with pressure-induced cell damage, friction, and shear it leads to ulceration in tissue that is already deep within the inflammatory cycle, even before the wound appears.15
Once a newly created wound appears there is a rapid increase in bacterial cell counts with production of multicomponent extracellular polymeric structures, and further mobilization and recruitment of more inflammatory cells into the wound site.17 The microcirculation also begins to shows signs of fibrosis, as indicated by excessive deposition of type VI collagen and decreasing tissue oxygen levels creating the first signs of hypoxia.19 The presence of fibrinogen and fibrin is common in chronic wound and researchers have hypothesized that they scavenger growth factors and other signal molecules involved in healing.60 At this point, a newly created wound having by-passed the hemostasis phase of healing is now firmly locked into a self-sustaining state of chronic inflammation.18, 19
All chronic wounds are similar in that each is characterized by one or more persistent inflammatory stimuli: repetitive trauma, ischemia, or low-grade bacterial contamination. Once the skin barrier is broken and bacterial colonization occurs, inflammatory molecules from bacteria such as endotoxin, platelet products such as transforming growth factor β (TGF-β) or fragments of extracellular matrix (ECM) molecules such as fibronectin stimulate proliferation of inflammatory cells (neutrophils and microphages) to enter the wound. These activated inflammatory cells secrete proinflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β). In acute wounds these cytokines are tightly controlled; in chronic wounds they are not. When compared to acute wounds, chronic wound fluid contains up to a 100-fold increase in TNFα and IL-1β. 1, 20 – 22, 57, 58 Approximately 2 weeks after chronic ulcers start to heal, cytokine levels begin to drop and approach the levels found in acute surgical wounds.23, 27
The proinflammatory cytokines (TNFα and IL-1β) also synthesize matrix metalloproteinase (MMP) and suppress tissue inhibitors of matrix metalloproteinase (TIMP).1, 21 The chronic wound environment has been proven to contain elevated protease (MMP) levels and decreased levels of protease inhibitors (TIMP), when compared to acute wounds.20-22, 24-26 Matrix metalloproteinases play an important role in all phases of wound healing by promoting cell migration, breaking down extracellular matrix, and remodeling. An imbalance in the microwound environment with increasing numbers of MMP’s and decreasing TIMP’s levels is associated with the degradation of collagen and growth factors and growth factor receptor sites as well as other vital components of the extracellular matrix.20-22, 24- 26 Once growth factors are degraded, communication between the various cells participating in the wound healing process stops and wound healing is delayed.59 As the inflammatory cycle is prolonged it amplifies the pro-inflammatory cytokine cascade leading to wound fluid which has been found to be absent of DNA synthesis. This is related to the low mitotic activity, excessive levels of inflammatory cytokines, high levels of proteases, and excessive reactive oxygen species found in chronic wound fluid which in turn results in senescent or mitotically incompetent cells .20, 28,68
Once the inflammatory cycle is prolonged, it creates a “vicious cycle” which is characteristic of all chronic wounds.1 The self-sustaining nature of this vicious cycle has far reaching effects. As the wound shifts from hypoxic to ischemic over time the lack of available oxygen for metabolism leads to anaerobic metabolism which reduces concentrations of adenosine triphosphate (ATP) resulting in metabolic acidosis. The reduction of ATP facilitates increased lactate with decreasing pH which excites nociceptors and produces activation of pH-sensitive ion channels, resulting in pain.81 Likewise, if there is sufficient oxygen for aerobic metabolism, then the by-product acid is metabolized into carbon monoxide and water, which is the final step of complete metabolism. As a result, pain is alleviated or disappears.29 – 37 This may accounts for why acute wounds have lower levels of associated pain when compared to chronic wounds.
Incorporating biofilm development into the model of chronic inflammation may better explain the biochemistry and cellular biology of their effect on perpetuating chronicity and preventing healing.56 For example, chronically elevated pro-inflammatory cytokines (TNFα and IL-1β) increased MMP levels (MMP-2, 8, and 9) and increased elastase can be explained by the possible effects of a biofilm on the host’s innate immune system.38, 56 Biofilms may also influence fibroblast senescence, keratinocyte impairment and failure of endothelial cell to initiate angiogenesis.38, 56 All wounds have bacteria on their surface and despite this, may heal.39 Clinicians often associate infection with a number greater than 105 of culturable bacteria in a wound. However, a clinical biofilm bacteria may not culture, but it is still viable.40 Because biofilm bacteria are encapsulated in a self-made matrix of biopolymers that offers structural stability and protection they may not initiate the potent inflammation response that other planktonic, highly virulent, bacteria may produce and may be overlooked using traditional sampling techniques.40-45 It seems within reason that host responses and protease activity may be related to biofilm formation in the chronic wound.56
Hypergranulation tissue is believed to occur as a result of an extended inflammatory response.46, 47 Few articles have been published on this subject with the majority of them coming from the veterinary community related to the care of equines. The predisposing factors for hypergranulation tissue formation in horses include: hypoxia, infection, trauma/pressure combined with a prolonged inflammatory reaction.48-51 The focus of this research had been on transforming growth factor-β (TGF-β) which is generally acknowledged to be a cytokine with the ability to retard or accelerate granulation tissue formation.52 One fact stands out: angiogenesis and granulation tissue formation are stimulated by local factors of the microenvironment including: fibroblast, macrophages, endothelial cells and growth factors, with their migration dictated by MMP’s.1 The concept of a prolonged inflammatory phase with amplified pro-inflammatory cytokine cascade leading to elevated protease activity and impaired growth factors function may account for the presence of hypergranulation tissue in chronic wounds.
When cells die by apoptosis the cell membrane remains intact, thus allowing them to be recognized and phagocytosed by macrophages. Phagocytosis is a major mechanism used to remove pathogens and cell debris from the wounds. But when cells die by necrosis, they burst and release cytotoxic compounds that prolong the inflammatory response.53 Acute wounds heal without infection because of the microbiocidal capacity of these macrophages.1 For phagocytosis to be optimized it is dependent on an adequate blood supply with sufficient amounts of oxygen.54 A restriction in oxygen availability reduces the ability of macrophages to function at capacity, and may allow the formation of biofilms in the presence of low grade infections. Macrophages ingest microorganisms and excrete products of digestion, namely ascorbic acid, hydrogen peroxide, and lactic acid as a result of phagocytosis. Hydrogen peroxide aids in controlling anaerobic microbial growth, while the levels of ascorbic and lactic acids are interpreted as a need for macrophages.54, 55 This increases the vicious cycle of chronic inflammation as more macrophages produce more by-products, leading to a more intense and prolonged inflammatory response, which diminishes the ability of macrophages to recognition and remove cells, thereby promoting necrotic disintegration and perpetuation chronic inflammation.56
Free radicals are known to cause cell damage and function as inhibitory factors in the healing process.61-63 As this is a complex process, only a brief overview of key points will be discussed. The production of reactive oxygen species (ROS) associated within a chronic wound can originate from several potential sources.65-68 During the healing process, various inflammatory cells; neutrophils, macrophages, endothelial cells, fibroblasts, and in particular senescent fibroblasts which are prominent in chronic wounds, are capable of produce superoxide.88-90 But, in chronic wounds activated neutrophils and macrophages produce extremely large amounts of superoxide and its derivatives via the phagocytic isoform of NADPH oxidases.69-70, 75 When polymorphonuclear neutrophils (PMNs) are recruited and activated at the wound site they consume an increased amount of oxygen, which is converted into ROS, in a process known as a “respiratory” or “oxidative” burst. This burst requires the consumption of large amounts of molecular oxygen, increasing oxygen consumption by at least 50%.64 Resulting in the generation of superoxide anions. Most of the superoxide anions formed are converted into hydrogen peroxide.71 Some of the hydrogen peroxide (H2O2) is converted into highly toxic hydroxyl radicals via the iron-catalysed Fenton reaction, creating a second source of ROS .72-74 Iron is released from hemoglobin by degraded erythrocytes, ferritin, and hemosiderin.76,82 High iron levels have been in the wound fluid of other types of chronic ulcers.66,82 An example of this is venous ulcerations which have excessive iron deposition in the skin, which is used in a Fenton reaction to produce excessive amounts of ROS, in this case hydroxyl radicals.77,82 These highly toxic hydroxyl free-radicals also enhance the synthesis and activation of even more matrix-degrading metalloproteinase.78-80,82,83
The presence of excessive reactive oxygen metabolites are not only highly toxic to surrounding tissue, they also increase MMPs while decreasing TIMP levels, creating a highly aggressive chronic wound environment that inhibits healing and deepens the chronic wounds inflammatory cycle. In addition, researchers have found that oxidative stress is also responsible for fibroblast senescents in chronic wounds.84-90
References
- Mast BE, Schultz GS. Interactions of cytokines, growth factors, and proteases in acute and chronic wound. Wound Repair and Regeneration. 1996; 4: 411-420.
- Coleridge Smith PD, Thomas P, Scurr JH, Dormandy JA. Causes of venous ulceration: a new hypothesis. BMJ 1988; 296: 1726-1727.
- Greenwood JE, Edwards AT, McCollum CN. The possible role of ischaemia-reperfusion in the pathogenesis of chronic venous ulceration. Wounds. 1995; 7: 211-219.
- Coleridge Smith PD. The microcirculation in venous hypertension. Cardiovascr Res 1996; 32: 789-795.
- Coleridge Smith PD. Oxygen, oxygen free radicals and reperfusion injury. Wounds.1996; 8: 9-15.
- Coleridge Smith PD. Neutrophil activation and mediators of inflammation in chronic venous insufficiency. J Vas Res. 1999; 36: 24-36.
- Kilgore KS, Todd III RF, Lucchesi BR. Reperfusion injury. In: Gallin JI, Snyderman R, editors. Inflammation: basic principles and clinical correlation. Lippincott, Williams & Wilkins, 1999: 1047-1060.
- Nixon J. The pathophysiology and etiology of pressure ulcers. In: Morrison M, editor. The prevention and treatment of pressure ulcers. Edinburgh: Mosby, 2001: 17-36.
- Fagrell B, Jorneskog G, Intaglietta M. Disturbed microvascular reactivity and shunting – a major cause for diabetic complications. Vasc Med 1999; 4: 125-127.
- Dinh TL, Veves A. Microcirculation of the diabetic foot. Curr Pharm Des 2005; 11: 2301-2309.
- Dinh TL, Veves A. A review of the mechanisms implicated in the pathogenesis of the diabetic foot. Int J Low Exterm Wounds 2005; 4: 154-159.
- Tooke JE, Brash PD. Microvascular aspects of diabetic foot disease. Disease. Diabet Med. 1996; 13 (Supp. 1): S26-S29.
- Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies. Plastic and Reconstructive Surgery. 2006; 117; 7S: 35S-41S.
- Menke NB, Ward KR, Tarynn M, Bonchey DG, Diegelmann RF. Impaired wound healing. Clinics in Dermatology. 2007; 25 (1): 19-25.
- Chen JWY, Rogers AA. Recent insights into the cause of chronic leg ulceration in venous diseases and implications on other types of chronic wounds. Wound Rep Reg. 2007; 15: 434-449.
- Tandara AA, Mustoe TA. Oxygen in wound healing – more than a nutrient. World Journal of Surgery. 2004; 28: 294-300.
- Mins CA. Mechanism of cell and tissue damage. In: The Pathogenesis of Infectious Disease. London: Academic Press, 1987: Chapter 8: 179-225.
- Stacey MC, Burnand KG, Bhogalm BS, Black MM. Pericapillary fibrin deposits and skin hypoxia precede the changes of lipodermatosclerosis in limbs at increased risk of developing a venous ulcer. Cardiovasc Surg 2000; 8: 372-380.
- Neumann HA, van den Brock MJ. Increased collagen VI layer in the basal membrane are of the capillaries in severe chronic venous venous insufficiency. Vasa 1991; 20: 26-29.
- Schultz G Mast BA: Molecular analysis of the environment of healing and chronic wounds: cytokines, proteases, and growth factors. Wounds 1998; 10: (Suppl F): 1F.
- Tranuzzer R. et al: Epidermal growth factor in wound healing: a model for the molecular pathogenesis of chronic wounds. In: Ziegler T, Pierce G, Herndon D, editors: Growth Factors and Wound Healing: Basic Science and Clinical Applications. Norwell, Mass, Springer. 1997
- Tranuzzer R, Schultz G. Biochemical analysis of acute and chronic wound environments. Wound Repair and Regeneration. 1996; 4: 321-3
- Harris I, et al. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp Dermatol. 1995; 4 (3): 42-
- Vaalamo M. et al. Patterns of matrix metalloproteinase and TIMP-1 expression in chronic and normally healing human cutaneous wounds. Br J Dermatol. 1996; 135 (1): 52-
- Wysocki A, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol. 1993; 101: 64-
- Yager D. et al. Wound fluids from human pressure ulcers contain elevated matrix metalloproteinase levels and activity compared to surgical wound fluid. J Invest Dermatol. 1996; 107: 743-
- Bullen E. et al. Tissue inhibitors of matrix metalloproteinase-1 is decreased and activated gelatinases are increased in chronic wounds. J Invest Dermatol. 1995; 104: 236-
- Telgenhoff D, Shroot B. Cellular senescence mechanisms in chronic wound healing. Cell Death and Differentiation. 2005; 12: 695-698.
- Kim TJ, Freml L, Park SS, Brennan TJ. Lactate concentrations in incisions indicate ischemic-like conditions may contribute to postoperative pain. The Journal of Pain. 2007; 8 (1): 59-66.
- Sutherland SP, Cook SP, McCleskey EW. Chemical mediators of pain due to tissue damage and ischemia. Prog Brain Res. 2000; 129: 21-38.
- Woo YC, Park SS, Subieta AR, Brennan TJ. Changes in tissue pH and temperature after incision indicate acidosis may contribute to postoperative pain. Anesthesiology. 2004; 101: 468-475.
- Reeh PW, Steen KH. Chapter 8. Tissue acidosis in nociception and pain. Progress in Brain Research. 1996; 113: 143-151.
- Steen KH, Reeh PW. Sustained graded pain and hyperalgesia from harmless experimental tissue acidosis in human skin. Neuroscience Letters. 1993; 154 (1-2): 113-116.
- Issberner U, Steen KH, Reeh PW. Pain due to tissue acidosis: a mechanism for inflammatory and ischemic myalgia? Neuroscience Letters. 1996; 208 (3): 191-116.
- Birklein F, Weber M, Neundörfer B. Increased skin lactate in complex regional pain syndrome: evidence for tissue hypoxia? Neurology. 2000; 55: 1213-1215.
- Naves LA, McCleskey EW. An acid-sensing ion channel that detects ischemic pain. Brazilian Journal of Medical and Biological Research. 2005; 38 (11): 1561-1569.
- Koban M, Leis S, Schultze-Mosgau S, Birklein Tissue hypoxia in complex regional pain syndrome. Pain 2003; 104 (1): 149-157.
- Wolcott RD, Rhoads DD. A study of biofilm based wound management in subjects with critical limb ischemia. Journal of Wound Care. 2008; 17 (4): 145-154.
- Dow G. Infection in chronic wounds. In: Krasner DL, Sibbald RG. Eds. Chronic Wound Care: A Clinical Source Book of Health Care Professionals. (3rd ed) HMP Communications. 2001.
- Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005; 13 (1): 34-40.
- Allesen-Holm, et al. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol. 2005; 59: 1114-1128.
- Klausen M. et al. Involvement of bacterial migration in the development of complex multicellular structures in pseudomonas aeruginosa biofilms. Mol Microbiol. 2003; 50: 61-68.
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999; 284 (5418): 1318-1322.
- Costerton JW, Veeh R, Shirtliff M, Pasmore M, et al. The application of biofilm science to study and control of chronic bacterial infections. Journal of Clinical Investigation. 2003; 112 (110): 1466-1477.
- Jensen ET, Kharazmi A, Garred P, et al. Complement activation by pseudomonas aeruginosa biofilms. Microb Pahog. 1993; 15: 377-388.
- Sato K, Komatsu N, Higashi N, Imai Y, Irimura T. Granulation tissue formation by nonspecific inflammatory agent occurs independently of macrophage galactose-type C-type lectin-1. Clinical Immunology 2005; 115 (1): 47-50.
- Shekhter AB, Berchenko GN, Nikolaev AV. Granulation tissue: inflammation and regeneration. Arkhiu Patologii. 1984; 46 (2): 20-29.
- Engelen M, Besche B, Lefay MP, Hare J, Vlaminck K. Effects of ketanserin on hypergranulation tissue formation, infection, and healing of equine lower limb wounds. Can Vet J. 2004; 45 (2):44-49.
- Berry DB, Sullins KE. Effects of topical application of antimicrobials and bandaging on healing and granulation tissue formation in wounds of the distal aspect of the limbs in horses. Am J Vet Res. 2003; 64 (1): 88-92.
- Theoret CL, Barber SM, Moyana TN, Gordon JR. Preliminary observations on expression of transforming growth factors beta 1 and beta 3 in equine full thickness skin wounds healing normally or with exuberant granulation tissue. Vet Surg. 2002; 31 (3): 266-273.
- De Martin I, Theoret CL. Spatial and temporal expression of type I and II receptors for transforming growth factor TGF-β; in normal equine skin and dermal wounds. Veterinary Surgery. 2004; 33 (1): 70-76.
- Roberts AB, Sporn MB. Transforming growth factor-β. In: Clark RAF. Ed. The molecular and cellular biology of wound repair. (2nd ed). Plenum Press 1988.
- Savill J. Apoptosis in resolution of inflammation. Journal of Leukocyte Biology. 1997; 16: 375-380.
- Sheffield PJ. Tissue oxygen measurements. In: Davis JC, Hunt TK. (eds) Problem Wounds: The Role of Oxygen. Elsevier. 1988.
- Hunt TK. Wound healing and wound infection: theory and surgical practice. Prentice-Hall. 1980.
- Wolcott RD, Rhoads DD, Dowd SE. Biofilms and chronic wound inflammation. Journal of Wound Care. 2008; 17 (8): 333-341.
- Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair and Regeneration. 1999; 7 (6): 433-441.
- Trengrove NJ, Langton SR, Stacey MC. Biochemical analysis of wound fluid from nonhealing and chronic leg ulcers. Wound Repair and Regeneration. 1996; 4 (2): 234-239.
- Ovington LG. Matrix metalloprotease modulation and growth factor protection. Podiatry Today. 2002; (Suppl.) 1-15.
- Falanga V, Grinnell F, Gilchrist B, Maddox YT, Moshell A. Workshop on the pathogenesis of chronic wounds. Journal of Investigative Dermatology 1994; 102: 125-127.
- Latha B, Babu M. The involvement of free radicals in burn injury: a review. Burns. 2001; 27 (4): 309-317.
- Thang PT, Patrick S, Telk LS, et al. Anti-oxidant effects of the extracts from the leaves of chromolaena odorata on human dermal fibroblasts and epidermal keratinocytes against hydrogen peroxide and hypoxanthine-xanthine oxidase induced damage. Burns 2001; 27 (4): 319-327.
- Galeano M, Torre V, Deodato B, et al. Raxofelast, a hydrophilic vitamin E-like antioxidant stimulates wound healing in genetically diabetic mice. Surgery 2001; 129 (4): 467-477.
- Rabkin JM, Hunt TK. Infections and oxygen. In: Davis JC, Hunt TK. (Eds) Problem Wounds: The Role of Oxygen. Elsevier. 1988.
- Widgerow AD. Deconstructing the chronic wound. Wound Healing South Africa. 2009; 2 (1): 09-11.
- Wenk J, Foitzik A, Achterberg V, et al. Selective pick up of increased iron by deferoxamine-coupled cellulose abrogates the iron-driven induction of matrix degrading metalloproteinase 1 and lipid peroxidation in human dermal fibroblasts in vitro: a new dressing concept. Journal of Investigative Dermatology 2001; 116: 833-839.
- Van den Berg AJJ, Halkes SBA, et al. A novel formulation of metal ions and citric acid reduces reactive oxygen species in vitro. Journal of Wound Care. 2003; 12 (10):
- Sonja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacological Reports. 2005; 57: (suppl.) 108-119.
- Babior BM, Curnutte JT, McMurrich BJ. The particulate superoxide forming system from human neutrophils. Properties of the system and further evidence supporting its participation in the respiratory burst. Journal of Clinical Investigation 1976; 58 (4): 989-996.
- Babior BM. Oxygen dependent microbial killing by phagocytes. N Eng J Med. 1978; 298 (12): 659-668.
- Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998; 92 (9): 3007-3017.
- Samuni A, Black CDV, Krishna CM, et al. Hydroxyl radical production by stimulated neutrophils reappraised. Journal of Biol Chem. 1988; 263 (27): 13797-13801.
- Harrison JE, Shultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem 1976; 251 (5): 1371-1374.
- Ackerman Z, Seidenbaum M, Loeweenthal E, Fukuto JM, et al. Overload of iron in the skin of patients with varicose ulcers. Possible contributing role of iron accumulation in progression of the disease. Arch Dermatol. 1988; 124: 1376-1378.
- Kalinowski L, Malinski T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: relationship to endothelial dysfunction. Acta Biochim Pol. 2004; 51: 459-469.
- Thomas CE, Morehouse LA, Aust SD. Ferritin and superoxide-dependent lipid peroxidation. J Mol Biol. 1985; 260: 3275-3280.
- Wenner A, Leu HJ, Spycher M, Brunner U. Ultrastructural changes of capillaries in chronic venous insufficiency. Expl Cell Biol. 1980; 48: 1-14.
- Saarialho-Kere UK. Patterns of matrix metalloproteinase and TIMP expression in chronic ulcers. Arch Dermatol Res. 1998; 290: S47-S54.
- Weckroth M, Vaheri A, Luharanta J, et al. Matrix metalloproteinase, gelatinase and collagenase, in chronic leg ulcers. J Invest Dematol. 1996; 106: 1119-1124.
- Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinase MMP-2 and MMP-9. J Invest Dematol. 1993;m101: 64-68.
- Kim TJ, Freml L, Park SS, Brennan TJ. Lactate concentrations in incisions indicate ischemic-like conditions may contribute to postoperative pain. Journal of Pain. 2007; 8 (1): 59-66.
- Yeoh-Ellerton S, Stacey MC. Iron and 8-isoprostance levels in acute and chronic wounds. Journal for Investigative Dermatology. 2003; 121: 918-925.
- Allhorn M, Lundqvist K, Schmidtchen A, Akerstrom B. Heme-scavenging role of α1-microglobulin in chronic ulcers. Journal for Investigative Dermatology. 2003; 121: 640-646.
- Clark RAF. Oxidative stress and “senescent” fibroblast in non-healing wounds as potential therapeutic targets. Journal for Investigative Dermatology. 2008; 128: 2361-2364.
- Stephens P, Cook H, Hilton J, Jones CJ, et al. An analysis of replicative senescence in dermal fibroblast derived from chronic leg wounds predicts that telomerase therapy would fail to reverse their disease-specific cellular and proteolytic phenotype. Exp Cell Res. 2003; 283: 22-35.
- Wall IB, Moseley R, Baird DM, Kipling D, et al. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. Journal for Investigative Dermatology. 2008; 128: 2526-2540.
- Song YS, Lee BY, Hwang ES. Distinct ROS and biochemical profiles in cells undergoing DNA damage-induced senescence and apoptosis. Mech Ageing Dev. 2005; 126: 580-590.
- Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. Journal for Investigative Dermatology. 2007; 127: 514-525.
- Campisi J. Replicative senescence: an old lives tale? Cell. 1996; 84: 497-500.
- Mendez MV, Stanley A, Parker HY, Shon K, Phillips T, Menzian JO. Fibroblasts cultured from venous ulcers display cellular characteristics of senescence. J Vas Surg. 1998; 28: 876-883.